Application:
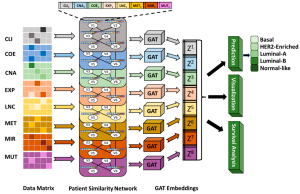
MOGAT
Accurate cancer subtype prediction is crucial for personalized medicine. Integrating multi-omics data represents a viable approach to comprehending the intricate pathophysiology of complex diseases like cancer. Conventional machine learning techniques are not ideal for analyzing the complex interrelationships among different categories of omics data. Numerous models have been suggested using graph-based learning to uncover veiled representations and network formations unique to distinct types of omics data to heighten predictions regarding cancers and characterize patients’ profiles, amongst other applications aimed at improving disease management in medical research. The existing graph-based state-of-the-art multi-omics integration approaches for cancer subtype prediction, MOGONET, and SUPREME, use a graph convolutional network (GCN), which fails to consider the level of importance of neighboring nodes on a particular node. To address this gap, we hypothesize that paying attention to each neighbor or providing appropriate weights to neighbors based on their importance might improve the cancer subtype prediction. The natural choice to determine the importance of each neighbor of a node in a graph is to explore the graph attention network (GAT). Here, we propose MOGAT, a novel multi-omics integration approach, leveraging GAT models that incorporate graph-based learning with an attention mechanism. MOGAT utilizes a multi-head attention mechanism to extract appropriate information for a specific sample by assigning unique attention coefficients to neighboring samples. Based on our knowledge, our group is the first to explore GAT in multi-omics integration for cancer subtype prediction. To evaluate the performance of MOGAT in predicting cancer subtypes, we explored two sets of breast cancer data from TCGA and METABRIC. Our proposed approach, MOGAT, outperforms MOGONET by 32% to 46% and SUPREME by 2% to 16% in cancer subtype prediction in different scenarios, supporting our hypothesis. Our results also showed that GAT embeddings provide a better prognosis in differentiating the high-risk group from the low-risk group than raw features.
CyFinder
CyFinder – A Cytoscape Plugin to Find Subgraph Biomarkers in Biological Networks (Github, Tutorial).
In CyFinder, we implemented graph theoretic and community detection algorithms to discover subnetwork biomarkers from biological networks, such as gene co-expression and protein-protein interaction networks. The CyFinder plugin comes with functionality such as finding the cliques and bipartite graphs, which could be the building blocks for disease progression at the protein network level. CyFinder offers a Bipartite Layout (not available in the Cytoscape) in the Layout Menu that visualizes the nodes of a bipartite network in two disjoint sets. We implemented three renowned community detection algorithms (Edge Betweenness, Fastgreedy, and Walktrap), unavailable in other plugins. CyFinder can find the intersection of two or more graphs, and the resulting graphs could help identify the common network biomarkers in a cohort of patients for a particular disease. We also implemented two minimum spanning tree algorithms to identify both minimum and maximum spanning forests from a weighted graph, which could help analyze disease pathways.
The CyFinder App is a outcome of NSF CAREER project titled “NetDA—Protein Network-Based Software for Disease Analysis Using Cliques, Bipartite Graphs, and Diffusion Kernels.”
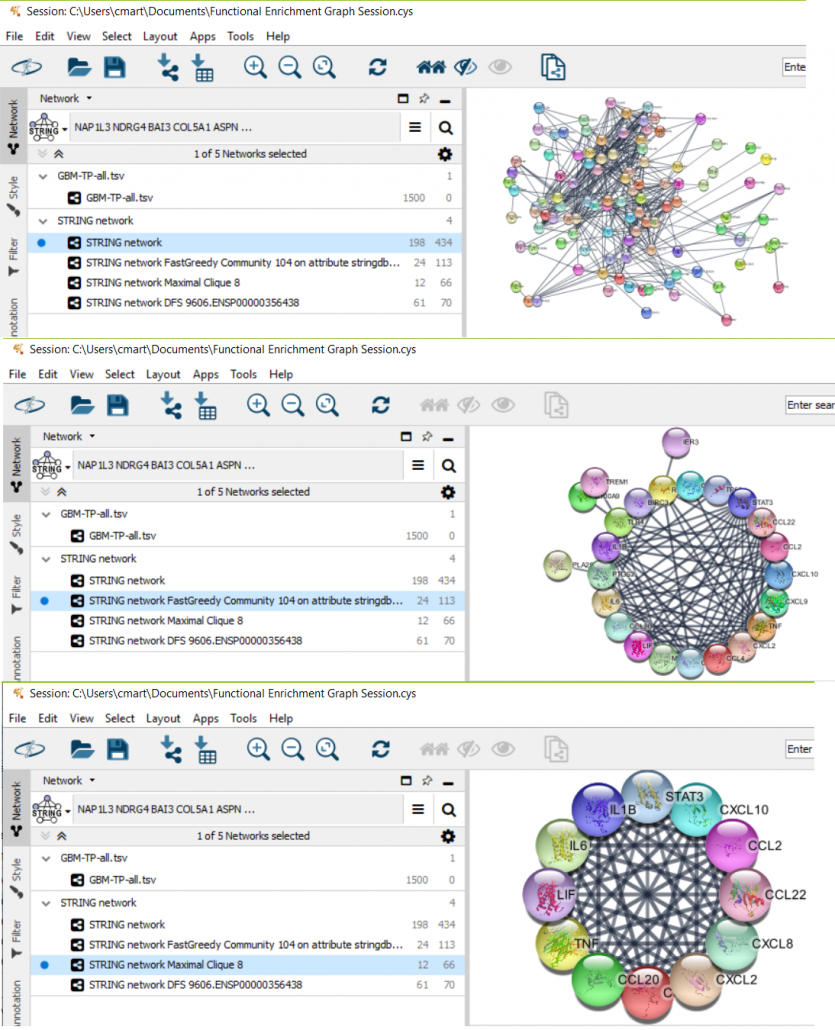
Sample CyFinder Results